



今回はNMR (Nuclear Magnetic Resonance, 核磁気共鳴)分光法です。主にこれが書きたくてこの復習ノートを書き始めたようなものです。これから研究室でNMRを読まなきゃいけないけど正直何が何だかわからない、というぐらいの人向けになります。
NMR の原理はMSやその他大多数の分析法の原理と比較しても格段に難しい説明になってしまうため、解説は別のページに譲ります。
サンプルの扱い方と溶媒の選定
現実的なセッティングと測定時間で十分な 1H-NMR のスペクトルを得るためには、十分に単離精製されたある程度の量の化合物が必要です。これは分子量100~500程度の低分子であれば一般に数mgです。
測定のためには、分析対象の化合物を何らかの溶媒に溶解する必要があります(固体NMRというわけのわからない技術もありますが、ここでは扱いません)。
この溶媒はNMRによって検出されない化合物である必要があります。すなわち、プロトン(1H)を含んでいてはいけないということです。もし 10-6~10-7 mol オーダー程度の微量な化合物を 1 mL のメタノールなんかに溶解して測定しようものなら、NMRはメタノールのプロトンのピークしか可視化してくれないことでしょう。
さて、そんな溶媒あったかな?
NMRの測定に供される溶媒は、一般的に測定専用の溶媒です。最もよく使われるのは CDCl3 ―クロロホルム-d6、重水素置換したクロロホルムです(研究室ではしばしば重クロ、と略して呼ばれています)。重水素で置換しているためプロトンのピークが出ず(CHCl3が不純物として検出されますがあまり他のピークを邪魔しない位置にきれいに出る)、比較的安価に製造可能であることから多用されています。クロロホルムに溶けない場合は CD3OD (メタノール-d4) や DMSO-d6、あるいはD2O (重水) などが使われますが、重水素が多い溶媒はその分高くつくのが常です。
また、内部標準物質としては普通 TMS (テトラメチルシラン) が用いられます。
過去に国試でも出たことがありますが、TMSはトリメチルシランではなくテトラメチルシランです。
構造を書いてみると、テトラメチルシランは (CH3)4Si と全てのプロトンが等価できれいな対称形であるのに対し、トリメチルシランは(CH3)3SiH と等価でないプロトンが存在することがわかります。
スペクトルの読み方
化学シフト
ざっくり言うと、それぞれのプロトンのスペクトルの化学シフトの値は、「近傍の原子団の電気陰性度の大きさ」と「磁気遮蔽効果の大きさ」によって定義されます。
学生当時、私は頭の中で「この核の周辺に電子がどれくらいありがちか」という妙な表現で理解していました(今も若干そう)。これだと磁気遮蔽効果を説明できていませんが、少なくとも近傍の原子団の電気陰性度について考える限りにおいては大体あってると思います。
有機化合物中のプロトンの化学シフトの一般的な領域は概ね以下のようになります。
| プロトンの位置 | 化学シフト | 備考 |
|---|---|---|
| 脂肪族、脂環式 | 0~2 ppm | |
| β-置換脂肪族 | 1~2 ppm | |
| アルキン | 2~3 ppm | |
| α-1置換脂肪族 | 2~5 ppm | |
| α-2置換脂肪族 | 2~7 ppm | せいぜい 6 ppmまで。7 ppm近いことは稀 |
| -OH | 4~5 ppm | シャープなピークではなく、ブロードする(横に大きく広がる) 溶媒を重水(D2O)にして測定するとピークが消失する(たまに国試で見る) |
| アルケン | 4~7.5 ppm | だいたい 4.5~6 ppmまで |
| 芳香族/複素環芳香族 | 6~9 ppm | だいたい 6.5~8 ppm周辺に集中する 国試でよく見る |
| R-CHO | 9~10 ppm | |
| R-COOH | 10~13 ppm |
高磁場側?
NMRスペクトルを読むときにしばしば高磁場側/低磁場側という表現を使いますが、これは以下のようになっています。
| 低磁場側 | ←→ | 高磁場側 |
| 化学シフト大 | ←→ | 化学シフト小 |
| スペクトルの左側 | ←→ | スペクトルの右側 |
慣れている人はさらっと言いますが、慣れていないと混乱しやすいのでよく覚えておきましょう。
多重度とカップリング定数(J)
あるプロトンのピークは、そのプロトンのスピンが近傍のプロトンのスピンとカップリングすることにより分裂します(原理についての説明はここでは省きます)。通常、ピークの形状に影響を与えるのはビシナルカップリング(3J)のケースです。
H-C-C-H
あるプロトンから3結合先にプロトンがいて互いにカップリングしているのがビシナルカップリング(3J)です。ジェミナルカップリング(2J)や遠隔カップリング(4J)もありますが、考慮すべきカップリングのほとんどは3Jです。
ピークが分裂した時のピークの先端の本数を多重度と呼びます。
- 1本:singlet, s (シングレット、カップリングしていない)
- 2本:doublet, d (ダブレット)
- 3本:triplet, t (トリプレット)
- 4本:quartet, q (カルテット)
以下同様に9本の場合まで規則に従って名前は付けられますが、実用的にはあまり意味はありません。またあまり細かく特定できない場合は単にmultiplet, m (マルチプレット)と表現します。
あるピークの多重度がnである時、そのプロトンの3結合先には化学的に等価なプロトンがn-1個ある、と覚えてください。キレられそうなくらい雑な説明ですが当分はこれだけで十分です。
実際に読んでみる
例として、イブプロフェンの1H-NMRスペクトルを見てみましょう。
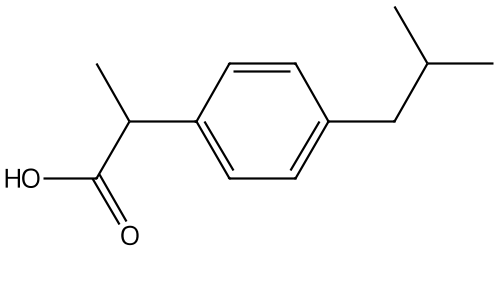
この化合物は分子式がC13H18O2で、観測可能なプロトンを18個持ちます。
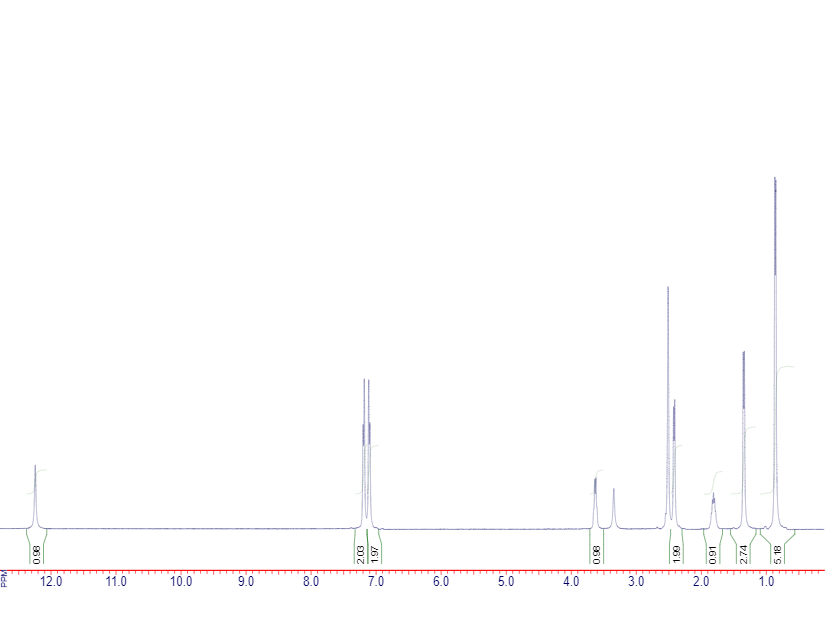
各ピークの下に書いてある数字は積分値といい、ピークの面積を表します。これはそのピークに対応するプロトンの数に比例します。化学的に等価なプロトン(例えば、1つのメチル基を構成している3つのプロトンはすべて等価です)は全て同じ場所にピークが出ますので、ここからどのようなプロトンがいくつあるのかを推定できます。また、実際に検出されるピークの数自体は18個よりは少なくなります。
ざっくりと以下のような感じでそれぞれのピークを評価します。化学シフト順ではなく、情報がすぐわかる順に上から並べています(これは私の読み方なので、人によっては順番は変わるかもしれません)。
| 化学シフト | 多重度 | 見立て |
|---|---|---|
| >12 ppm | s | ほぼカルボン酸 |
| 7.1~7.3 ppm | dd | この辺のピークは芳香族。 積分値4かつdd(ダブルダブレット)なのでパラ-2置換の芳香族化合物 |
| 0.9 ppm | d | 雑なメチル 積分値5.18だけどこれは6な気がする(この辺りは経験に基づく勘です。メチル基なら3の倍数のはずです) d(ダブレット)なのでおそらくR-CH-(CH3)2 |
| 1.8 ppm | m | ちょうどいいところにm(マルチプレット/よくわからない)で積分値1のピークがあるのでたぶんこれがR-CH-(CH3)2 |
| 2.4 ppm | d | 積分値2、dなのでR1-CH-CH2-R2 |
| 3.6 ppm | q | おそらく-COOHの近所にあるために低磁場側に引っ張られているピーク ※多重度は一見dに見えるがキレイなスペクトルならqであることがはっきりわかる |
| 1.3 ppm | d | 積分値2.74はたぶん3なのでメチル、dなのでR1R2CH-CH3? →3.6 ppmのメチルがついているプロトンがあるので、R-CH(COOH)(CH3)ということになりそう かつこのRはCHnではない(もしそうならマルチプレットになる)から、1.8 ppmのプロトンはR1-CH2-CH-(CH3)2の形になる |
ということで(あとは以上の情報を統合するだけですので割愛しますが)、なんと分子式と1H-NMRスペクトルだけでイブプロフェンの同定に成功しました。論文に書く時はこんな書き方したらブチギレられますが、頭の中で考えてることなんて正味こんなもんです。
なお、積分値の書いていないピークがありますが、これは溶媒の不純物などのピークです。
いつもこれくらいの簡単な化合物なら良いですが、実際には情報がこれだけでは大抵の有機化合物の構造の同定は難しいので、さらに結合定数(J)の情報なども加味して様々な化合物の同定に利用します。
次回は13C-NMRについてみていきます。


